Uterine endometrial cancer incidence (~63,200 new cases/year) and deaths (~11,300 deaths/year) are on the rise. This tumor type is exquisitely sensitive to the growth promoting effects of estrogen and the growth limiting effects of progesterone. Hormone therapy using progesterone or synthetic analogues, progestin, for endometrial cancer has been a traditional choice for treatment by inducing differentiation, promoting apoptosis, and inhibiting invasion. It is highly effective in the short term; however, responsiveness wanes over time due to loss of progesterone receptor (PR) expression, and recurrences are common.
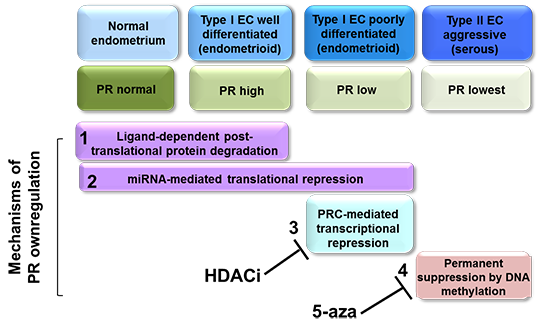
Our group has systematically dissected the mechanisms by which PR is lost and demonstrated that PR expression can be downregulated at four distinct molecular levels during disease progression (PMID: 25229191): 1) coarse post-translational regulation, mediated by ligand-dependent PR activation and downregulation ; 2) translational regulation, implemented by miRNAs that fine-tune PR expression; 3) transcriptional regulation, achieved by the repression of PR transcription by the Polycomb Repressor Complex (PRC2); and 4) permanent transcriptional repression of PR by DNA methylation. Levels 1 and 2 are reversible and occur under physiological conditions. Levels 3 and 4 are irreversible and occur under pathological conditions.
The long-term goal of my research is to develop evidence-based therapeutic interventions to improve survival for endometrial cancer patients as well as other hormone-driven cancer patients.
To achieve this goal, six projects will be conducted in my laboratory.
Project 1: Restoring Progestin Sensitivity in Endometrioid Endometrial Cancer.
The objective of this project is to identify the molecular mechanisms driving the downregulation of the progesterone receptor (PR) in endometrial cancer patients and identify novel strategies to further enhance the effectiveness of progestin therapy. My group has led efforts to understand the mechanisms underlying loss of PR in endometrial tumors. For example, we identified the significant improvement in progestin effectiveness when a histone deacetylase inhibitor (HDACi) is combined with progestin therapy, leading to the approval of a new NIH NCTN trial, NRG-GY011. GY011 is a surgical window trial, the first in humans, to test the hypothesis generated by our laboratory that an HDACi de-represses PR silencing in endometrial cancer. To maximize clinical interventions, we have now identified novel small molecule PR inducers as well as additional PR suppressors that have the potential to more clearly define the multiple mechanisms of PR inhibition in endometrial cancer, setting the stage for new therapeutic opportunities.
Project 2: Novel drug combinations to enhance hormone therapy.
We will validate new PR inducers and to test it in vitro and in vivo assays for its effect on increasing PR expression and inhibit endometrial tumor growth.
Project 3: Investigating epigenetic therapies in advanced, persistent or recurrent endometrial cancer.
Compare with endometrioid endometrial cancer, PR expression is lowest in advanced endometrial cancer. Progestin therapy also has lowest response rate for this type of endometrial cancer. Our previous data demonstrated that PR expression can be restored by hypomethylating agent, 5-aza-decitabine. Combination of HDACi with hypomethylation agent will restore PR expression dramatically in many endometrial cancer cells. We will apply this treatment strategy in advanced endometrial cancer.
Additional Projects
Moreover, several new projects will be conducted in our laboratory:
- Determine anti-tumor immune response in MPA+HDACi treatment;
- To determine the mechanism of PR as a negative transcriptional regulator for oncogene Myc in endometrial cancer;
- To extend molecularly enhanced progestin therapy to other cancer types.